
Tumor Suppressor Genes
Multiple Endocrine Neoplasia Type 1 (MEN1)
Introduction
Initiation and progression of cancer is driven by an increase in the activity of oncogenes that drive cell growth, and/or inactivation of tumor suppressor genes (TSGs) that slow or stop cell growth. TSGs represent some of the most well-known and iconic cancer-related genes, such as those responsible for retinoblastoma, inherited breast cancer, and others.
Of particular interest to Avoneaux Med is the Multiple Endocrine Neoplasia Type 1 (MEN1) gene, a TSG that causes a classic inherited multi-tumor syndrome that was first described in the 1950s.
MEN1 is an autosomal dominant syndrome of parathyroid, pancreatic, gastroduodenal, and pituitary gland neoplasms, collectively called neuroendocrine tumors (NETs). Surgical resection remains an effective treatment for localized NETs; however, for disseminated tumors surgery provides only limited survival benefit and is reserved for palliation. Similarly, chemotherapy and radiation treatment are minimally useful for reducing mortality in patients with metastatic disease, although some agents provide symptom relief by reducing tumor hormone release, e.g., somatostatin analogs.
Clearly, there is a need for more efficacious treatment of disseminated NETs to improve survival, both in patients with familial cancer and in the much larger group who develop counterpart sporadic tumors.
Gene Identification
The MEN1 gene was discovered by the NIH MEN1 working group in Bethesda, MD in 1997. Subsequent investigation found the gene was mutated in the germline in approximately 80% of affected MEN1 kindreds and inactivated via a classic two-hit mechanism in a significant fraction of both familial and sporadic NETs, contributing to a large public health burden.
The NIH MEN1 team was comprised of multiple Institutes and a diverse group of investigators, which was critical to the success of the effort. The overall project was led by Drs. Francis Collins and Settara Chandrasekharappa, with significant support from Judy Crabtree, SC Guru, Manickam Pachiappan, SE Olefumi, Y Wang of NHGRI; Stephen Marx, Allen Spiegel, Mary Beth Kester, Christina Heppner, Qihan Dong, YS Kim, Lee Burns, and Sunita Agarwal of NIDDK; Mark Boguski, Jane Weisemann, and David Lipman of NCBI; and Zhengping Zhuang, Irina Lubensky, Larisa Debelenko, Lance Liotta, and Steven Libutti of NCI.
Transcription Factor Model
Shortly after gene cloning, studies from the NIH MEN1 team using in vitro cell line models indicated the gene product menin is a nuclear protein that regulates mRNA transcription. Taken together with work performed by other groups, the transcription factor model of the MEN1 gene is now a well-established orthodoxy in the field and underlies several neuroendocrine tumor drug development programs.
Unfortunately, in the 25+ years since gene discovery no successful anti-NET drugs based on the transcription factor model were developed and no such agents are routinely used today. Thus, it’s important the field continually reexamine the premise that menin causes NETS via a role as a transcription factor.
(Note that some laboratories are currently using MEN1 gene fusions and other menin protein interactions as therapeutic targets in leukemia. While this is an interesting biological approach and promising clinical strategy, these fusions and protein interactions do not appear related to endocrine cancer and are not currently a target for anti-NET drug development.)
A Key Mystery for MEN1 and Other Inherited Cancer Syndromes
An important biological question that remains unanswered for the MEN1 gene and for many TSGs is why are DNA germline mutations present in all cells in the body, yet tumors arise only in specific and unique organs in each syndrome? Moreover, why are TSG mRNAs and proteins expressed in many cell types including the vast number that do not develop cancer?
For anti-cancer drug development it is essential to know the answer to the mystery since one wants to restore the tumor-specific function of a TSG to treat patients. Otherwise, significant resources are devoted to creating drugs for the wrong biological purpose.
A simple explanation is that TSGs have two biological functions; an older, ancestral constitutional role that occurs in all cells, and a more recently evolved specialized activity that is associated with cell growth. The latter function is present only in tissues that give rise to tumors, e.g., endocrine pancreas in MEN1, breast/ovarian in BRCA1, kidney in VHL, and so forth.
Thus, as a general matter there are critical research issues to tackle after discovery of a novel TSG: What is unique about the status of the gene and its protein product in tumor target cells versus other cells? What is the mechanism of this restricted tumor development? Are there differences in gene regulation between the constitutional function and the tumor-specific role? Is the TSG protein processed differently in tumor cells? Is there a unique TSG binding partner in tumor target cells?
Unexpected Menin Finding
After discovery of the MEN1 gene, menin expression was studied across a range of human surgical and autopsy tissues using immunohistochemistry (IHC) to assess menin in target organs and cells versus other tissues.
Something quite provocative was discovered.
Menin was found in the nucleus of all cells in the body, consistent with the prediction from the cell line models that the protein is a transcription factor. However, menin immunoreactivity was also highly and uniquely expressed in the cytoplasm of islet cells in human pancreas specimens, indicating the protein might have a unique role in this MEN1 target organ. The Figure below shows IHC evaluation of pancreas using an anti-menin antibody at the light microscope level (panels A, B, D-F). Subsequent immuno-electron microscopy studies localized the staining specifically to secretory granules (panel C), and an immunoblot against pancreatic tissue using the antibodies showed high specificity for menin at 63 kDa (panel G). Nuclear menin expression was also present in pancreatic endocrine and exocrine cell nuclei (expression is too low to be observed in the figure due to the IHC conditions utilized). In contrast, the secretory granule immunoreactivity in endocrine pancreas was expressed at high levels and readily observed.
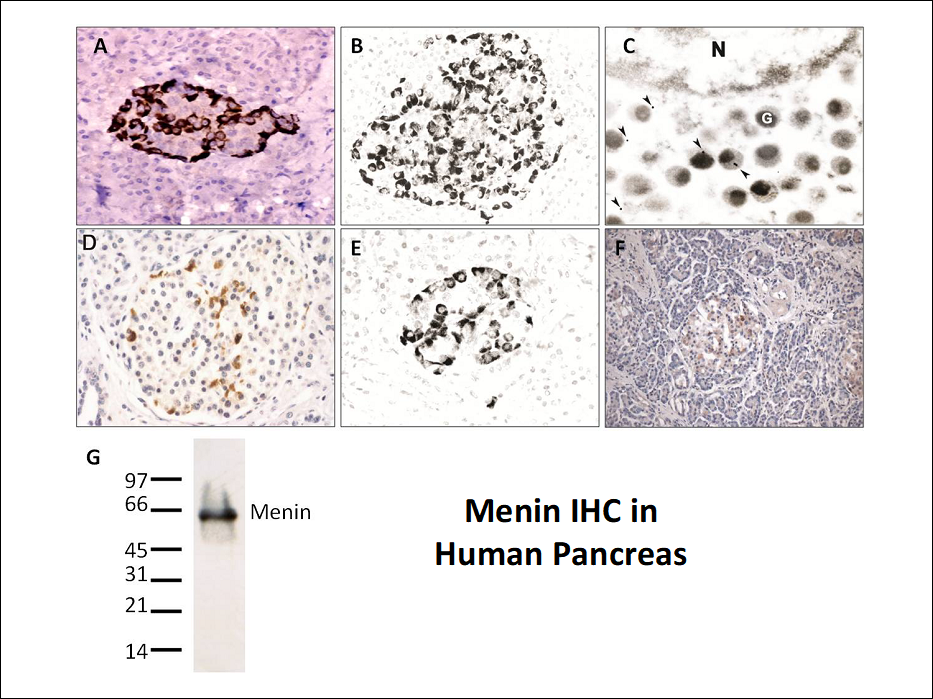
Most genetic and biochemical studies of the MEN1 gene and menin are/were performed in non-endocrine, non-tumor target systems since standard, off-the-shelf cultured cell lines and reagents are readily available and easy to manipulate (e.g., gene transfection studies). However, by definition these studies focus on the potential constitutional role that the gene/protein plays since the experimental systems are not endocrine cells. Thus, the conclusion that menin is a nuclear transcription factor, which is now dogmatic in the field, may be true for a constitutional role in all cells, but irrelevant to neuroendocrine tumor formation where the protein has a unique biological role.
IHC is a powerful and valuable technique; however, there are several technical pitfalls that accompany immunostaining of tissue. Thus an intensive effort was undertaken to confirm or falsify the anti-menin immunoreactivity finding. Six antibodies raised against full-length menin or its peptides were created and studied in pancreatic tissue from MEN1 patients and normal controls. The studies confirmed anti-menin immunoreactivity in secretory granules of endocrine pancreas. The staining was robust and reproducible and identical staining occurred with two separate anti-menin antibodies. Moreover, menin immunoreactivity was lost in NETs with MEN1 gene mutations as predicted by two-hit gene inactivation (evaluated using IHC and immunoblots in normal islets and tumors), and the observed endocrine pancreas staining was not due to a known islet hormone (e.g., insulin, glucagon). Additionally, the specific menin epitope targeted by the antibodies was identified and IHC staining was blocked using excess peptide specific to the epitope.
Although these efforts are consistent with menin being responsible for the unique staining in secretory granules in pancreatic islet cells, they do not definitively prove it to be true. There is some non-zero possibility the staining is due to antibody cross-reaction with a protein that shares a similar epitope with menin. Therefore, when these results were published to alert the field to the provocative finding, the observed staining was referred to only as “anti-menin immunoreactivity” not definitively as menin.
Potential Novel Paradigm and Therapeutic for NETs
If the observed IHC staining in pancreatic cells is indeed menin, then a simple model that explains both the in vitro cell line data (nuclear expression in all cell types) and the patient-based IHC tissue section data (secretory granule expression uniquely in endocrine pancreas) is that menin has a constitutional transcription factor function in all cells throughout the body, but a unique cancer-associated role in secretory granules of endocrine cells.
One can propose menin is an autocrine (or locally acting paracrine) negative growth regulator in endocrine pancreas that is released into the extracellular milieu, binds to a receptor on the outer surface of the releasing cell (and/or nearby cells), and suppresses growth via a negative feedback loop. Inactivation of the MEN1 gene and loss of menin expression eliminates the negative growth regulation and produces focal islet cell hyperplasia followed by frank NET formation. There is precedent for autocrine and local paracrine feedback controlling secretion/growth in many neural and endocrine cell types, including in pancreatic islets.
Based on the model a new therapeutic strategy emerges to treat disseminated NETs. Menin or the active small peptide within the protein can be administered exogenously to patients as a drug to replace the menin that is lost due to two-hit MEN1 gene inactivation. The protein/peptide will bind to the outer surface of tumor cells, induce the negative growth feedback loop that (now missing) endogenous menin normally activates, and slow/stop tumor growth. The drug could have utility for MEN1 patients and for the much larger number of patients with sporadic NETs with MEN1 gene mutations.
Further work by our group and others will be necessary to unravel the mystery of the observed anti-menin immunoreactivity in endocrine pancreas. Certainly, challenging the dogma of the transcriptional role of menin in endocrine tumorigenesis should be undertaken as part of the normal course of healthy scientific skepticism, even if the transcriptional model is ultimately proven true.
Selected References
- Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997 Apr 18;276(5311):404-7. doi: 10.1126/science.276.5311.404. PMID: 9103196.
- Localization of the multiple endocrine neoplasia type I (MEN1) gene based on tumor loss of heterozygosity analysis. Cancer Res. 1997 May 15;57(10):1855-8. PMID: 9157974.
- Haplotype analysis defines a minimal interval for the multiple endocrine neoplasia type 1 (MEN1) gene. Cancer Res.1997 Mar 15;57(6):1039-42. PMID: 9067266.
- Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997 Aug;16(4):375-8. doi: 10.1038/ng0897-375. PMID: 9241276.
- A transcript map for the 2.8-Mb region containing the multiple endocrine neoplasia type 1 locus. Genome Res. 1997 Jul;7(7):725-35. doi: 10.1101/gr.7.7.725. PMID: 9253601; PMCID: PMC310681.
- 11q13 allelotype analysis in 27 northern American MEN1 kindreds identifies two distinct founder chromosomes. Mol Genet Metab. 1998 Feb;63(2):151-5. doi: 10.1006/mgme.1997.2649. PMID: 9562970.
- A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001 Jan 30;98(3):1118-23. doi: 10.1073/pnas.98.3.1118. PMID: 11158604; PMCID: PMC14718.
- Menin immunoreactivity in secretory granules of human pancreatic islet cells. Appl Immunohistochem Mol Morphol. 2014 Nov-Dec;22(10):748-55. doi: 10.1097/PAI.0000000000000046. PMID: 25153502; PMCID: PMC4232475.